Additional Features¶
This chapter discusses the latest additions to TROVE and their implementation. Many of these different features are still being tested and developed and so this chapter, even more than others, should be treated as subject to change. These features have already proven important however and so will likely remain a part of TROVE but perhaps with a different implementation.
Most of these features are for dealing with larger molecules in TROVE.
Splitting and Stitching¶
As discussed in Chapter Quickstart, matrix elements between contracted basis functions are saved to a file, contr_matelem.chk. This step is time consuming and for large molecules/basis sets it may not be possible to carry out this step within a computer’s allocated time limit. The split command solves this problem by splitting the matrix into 12 ‘slices’. Each slice contains matrix elements of a component of the full Hamiltonian operator.
To use splitting, in the CHECK_POINT block the matelem should be set to
matelem save split i j
where i and j are integers between 0 and 12 (and j 
i). The most parallel way of saving the matelem files is to calculate each separately by setting i and j to 0 0, 1 1, …12 12. Splitting can also be used for the dipole using
extmatelem save split i j
where i and j are now 1,2 or 3 and should be run with 1 1, 2 2, 3 3. Using splitting will produce files matelemI.chk where i is 1-12 and extmatelemJ.chk where J
is 1-3.
To convert these files to the J=0 representation, use
matelem convert split
This will produce j0_matelemI.chk files. This can be used in subsequent calculations using read as usual, as long as split is specified also so that the keyword should be set to
::
matelem read split
end{verse}
Matrix elements of the dipole are converted in the same way. For the dipole it may be necessary to combine the split extmatelem files however using the stitch option as currently the GAIN program does not accept split files. To carry this out, after the three extmatelem files have been calculated, re-run TROVE with
extmatelem convert stitch
This will combine the split matelem files into a j0_extfield.chk files (which are produced if not splitting is carried out as specified in Chapter Quickstart). This can then be used with GAIN.
Restarting¶
For large molecules and basis sets it may be that the vibrational part of the calculation does not finish within a large computer’s time limit. In this case it is usually the calculation of matrix elements which is the time consuming part. Since these are independent calculations however, the elements can be saved and read back in to form the full Hamiltonian matrix.
To use this restart feature, in the CHECK_POINT block the matelem keyword should be set to
matelem save split 0 0 dump
In this case the dump keyword gets TROVE to sequentially save the matrix at intervals. This will produce an output file which after giving the dimensions of the contracted matrix will print out to which matrix element TROVE has saved. If the calculation then runs out of time, the user can read back in these pre-computed elements up to that point by specifying
matelem append n split 0 0 dump
In this case TROVE restarts at n and continue to calculate matrix elements. This can be repeated until the matrix is filled and be diagonalized.
Again, this is only for the 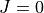 vibrational step and is useful when used in conjunction with the
vibrational step and is useful when used in conjunction with the J=0 step and especially with pruning methods, as described below.
Fast-CI¶
The Fast-CI method was implemented in TROVE by Andrey Yachmenev as a way of speeding up calculations. The method does this by essentially re-ordering loops and indices in a more efficient manner. Another aspect of the method is the ability to choose a cut-off where expansion coefficients of the Hamiltonian are discarded if too small. The method is optimally designed when using curvilinear coordinates.
To use the Fast-CI method, in the CONTRACTION block put fast_ci. To make use of the cut-off option put exp_coeff_thresh in the same block of input followed by what value is required. Typically very small values such as  or so should be used. This should result in essentially the same results as not using any cut-off but with significant speed up. If even faster calculations are required the value should be increased but values larger than
or so should be used. This should result in essentially the same results as not using any cut-off but with significant speed up. If even faster calculations are required the value should be increased but values larger than  are not recommended. Ideally this parameter should be benchmarked using smaller basis sets to get an idea of the particular systems sensitivity to its value.
are not recommended. Ideally this parameter should be benchmarked using smaller basis sets to get an idea of the particular systems sensitivity to its value.
The Fast-CI method generates different checkpoint files for the vibrational matrix elements compared to what is produced using standard TROVE options. Many files starting with contrME_ are produced which have to do with the ordering procedure.
When using Fast-CI, TROVE can be used as normal for other steps in the calculation and the fast_ci and exp_coeff_thresh keywords can remain the the CONTRACTION block.
Storing Hamiltonian and External Diagonalization¶
For larger production calculations involving high values of  it is likely that the construction and diagonalization of the Hamiltonian matrix will exceed the time limit of the computer which is used. In this case the Hamiltonian matrix can be saved and then diagonalized separately for each symmetry.
it is likely that the construction and diagonalization of the Hamiltonian matrix will exceed the time limit of the computer which is used. In this case the Hamiltonian matrix can be saved and then diagonalized separately for each symmetry.
To save the Hamiltonian for a given symmetry, in the DIAGONALIZER block put
save
gamma i
where i is the symmetry. This should be done for each symmetry at a given . TROVE will produce the files matrixJ_I.chk where J is the value of and I is the symmetry. TROVE will also produce eigenvector and descriptor files but these will be empty.
These matrices can then be diagonalized using an external program. An example of such a program is PDSYEVD, a ScaLapack program which is MPI parallel and so can be run on multiple CPU nodes. A TROVE compatible driver program for this is available from Sergey Yurchenko. An example input for this program is
(title)
J 32
gamma 8
DIAGONALIZER pdsyevd
ENERGY_THRESH 16000.0
COEFF_THRESH 1e-18
ZPE 11022.4701
MEM 64 gb
where the keywords are the same as those used in TROVE input.
This program will produce the eigenvectors for the specified and  as TROVE would but not the descriptor files (since only the Hamiltonian matrix was specified without details of the basis set, etc). The program also produces a
as TROVE would but not the descriptor files (since only the Hamiltonian matrix was specified without details of the basis set, etc). The program also produces a energiesJ_I.chk file which contains the eigenvalues.
To produce the relevant descriptor files and usual TROVE output files, TROVE should be re-run with the energiesJ_I.chk file in the same directory with the keywords in the DIAGONALIZER block changed to
read-energies
gamma i
This will cause TROVE to read the energies file and produce the usual descriptor files and output block containing the energies, quantum numbers, etc. This is essentially a ‘bookkeeping’ step and does not require much computing time or memory.
Transition Moment Intensity Pruning¶
Another method which has been developed to reduce calculation time of line lists for large molecules is transition moment intensity pruning. This procedure reduces how many vibrational levels are included based on their intensity. Levels which have very weak intensities for both transitions to and from them are discarded. This results in a large reduction of the basis set but should only remove transitions of very low intensity.
As the method prunes the basis using the vibrational intensities, it assumes that rotational levels with the same vibrational quantum numbers will also be weak.
To use this method, the usual steps for calculating the transition moment should be followed but the keyword pruning should be added into the intensity block. TROVE will then calculate the transition moments and intensities as usual but also work out and store the most intense transitions to and from each state. This calculations produces the checkpoint files eigen_intens0_n.chk for each symmetry n. In applications the temperature for this step has been set to around the maximum for which the line list being calculated to try and make sure no important states are left out.
The basis set can then be pruned using the J=0 method. In the CONTRACTION the following should be included
tm_cutoff 1e-24
tm_enermin 8000.0
tm_cutoff sets the minimum intensity for removing states. This should ideally by set to as low a value as possible and will depend on practical considerations such as computing time and memory. tm_enermin is the minimum energy in wavenumbers for which pruning will occur. In this example, all states below 8000 cm-1 will be included in the basis set regardless of intensity. This value should be set as large as possible but will again be determined by practical
considerations.
An example of using this procedure is for the ethylene (C2`H:sub:`4) molecule [18MaYaTe]. For this relatively large molecule a basis set
with a polyad number of 10 produced split matelem files which were 158 Gb each. Using these basis sets would not have been practical at high . Using the pruning method with the parameters as given in the example above reduced the matelems to 1.4 Gb. This then allowed refinement and a full line list calculation to be carried out.