Refinement¶
In this chapter details will be given of the refinement procedure implemented in TROVE. Refinement in this context means to adjust the ab intio potential energy surface by comparing computed rotational-vibrational energy levels to experimental values. Parameters of the PES are varied, energy levels re-computed and compared to experiment. This process is continued until acceptable agreement between the calculated and experimental energy levels is obtained. Usually there is relatively few experimental energies and so ab intio electronic energies are used to constrain the refinement to prevent over fitting. Although experimental data is usually at fairly low energies, it is often the case that correcting the lower energy region of the PES gives more accurate values at high energies also.
Refinement with TROVE: Theory¶
Details of the method of refinement implemented in TROVE have been published [11YuBaTe] and only a brief summary will be given here. Assuming a reasonable PES has
already been obtained, a correction is added in terms of internal coordinates  .. math:
.. math:
\Delta V = \sum_{ijk...} \Delta f_{ijk...} \left(\xi_1^i \xi_2^j \xi_3^k ...\right)^A
where  corresponds to totally symmetric permutation of the internal coordinates
so that all symmetry properties of the molecule are properly accounted for.
corresponds to totally symmetric permutation of the internal coordinates
so that all symmetry properties of the molecule are properly accounted for.  are the expansion coefficients which are found by refinement.
The Hamiltonian is now given as
.. math:
are the expansion coefficients which are found by refinement.
The Hamiltonian is now given as
.. math:
H = T + V + \Delta V = H_0 + \sum_{ijk...} \Delta f_{ijk...} \left(\xi_1^i \xi_2^j \xi_3^k \right)^A
where  is the initial Hamiltonian with ab initio PES.
is the initial Hamiltonian with ab initio PES.
If the eigenvalue problem for the initial Hamiltonian has been solved, .. math:
H_0 \psi^{J,\Gamma}_{0,i} = E^{J,\Gamma}_{0,i} \psi^{J,\Gamma}_{0,i}
where  is the total angular momentum quantum number and
is the total angular momentum quantum number and  is a symmetry label, then matrix elements of
is a symmetry label, then matrix elements of  ,
using the solutions as a basis, are
.. math:
,
using the solutions as a basis, are
.. math:
\left< \psi^{J,\Gamma}_{0,i} | H |\psi^{J,\Gamma}_{0,i'} \right> = E^{J,\Gamma}_{0,i} + \sum_{ijk...} \Delta f_{ijk...} \Xi_{i,i'}^{J, \Gamma}
where .. math:
\Xi_{i,i'}^{J, \Gamma} = \left< \psi^{J,\Gamma}_{0,i} | \left(\xi_1^i \xi_2^j \xi_3^k ...\right)^A | \psi^{J,\Gamma}_{0,i'} \right>.
The derivatives of the energies with respect to adjustable parameters, which are requred for least squares fitting, are given by the Hellman-Feynman theorem .. math:
\frac{\partial E^{J,\Gamma}_{n} }{ \partial \Delta f_{ijk...} } = \left< \psi^{J,\Gamma}_{n} \left| \frac{\partial \Delta V}{\partial \Delta f_{ijk...} } \right |\psi^{J,\Gamma}_{n} \right> = \left< \psi^{J,\Gamma}_{n} \left| \left(\xi_1^i \xi_2^j \xi_3^k ...\right)^A \right| \psi^{J,\Gamma}_{n} \right>.
where  and
and  are eigenvalues and eigenvectors of respectively.
In the J=0 representation is given by
.. math:
are eigenvalues and eigenvectors of respectively.
In the J=0 representation is given by
.. math:
\psi^{J,\Gamma}_{n} = \sum_i C_i^{J, \Gamma} \psi_{0,i}^{J, \gamma}
As the derivative of the energy levels with respect to the correction parameters are given, standard least squares fitting procedures can then be used to determine how they should be varied. This is all implemented in TROVE.
Refinement Implementation with TROVE¶
Setting up Refinement¶
The specific inputs and checkpoint files required to carry out refinement of a PES using TROVE is discussed in this section
Prior to refinement TROVE requires checkpoint files and eigenfunctions for the basis set being used (see above). If a calculation of the rotational-vibrational levels using an unrefined PES has already been carried out, then all necessary files for refinement will have been generated. Refinement can be carried out in the 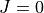 basis.
basis.
The refinement parameters ( from previous section) are defined in the external` block on the TROVE input file. If the PES to be refined is of the same form, that is, in terms of a polynomial of symmetrised internal coordinates, then the refinement parameters will just be a repeat of
the potential block.
from previous section) are defined in the external` block on the TROVE input file. If the PES to be refined is of the same form, that is, in terms of a polynomial of symmetrised internal coordinates, then the refinement parameters will just be a repeat of
the potential block.
The required matrix elements of the refinement parameters are computed in stages. First matrix elements of the primitive basis functions used by TROVE are calculated. This is carried out by putting
extmatelem save split n n
in the TROVE input file in the checkpoint block. n is the number of a specific expansion parameter. If all parameters are required this can be ignored but this may not be possible for all molecules or if there are lots of expansion parameters. This will generate extmatelemn.chk files (where n is number of expansion parameter). These files can then be converted to the J=0` representation using
extmatelem convert split n n.
As discussed above, the refinement procedure requires matrix elements of the Hamiltonian and so eigenfunctions for each of interest must be computed. To save matrix elements of the eigenfunctions, TROVE is run for each with
fit_poten save split
in the checkpoint block. This generates fitpot-J-Gamma-n.chk files. Since a file is
generated for each and symmetry for each expansion parameter n, many files are generated in this step.
Running Refinement¶
When all of the required checkpoint files described above have been generated, TROVE can be used to refine the PES. The following block should be added to the TROVE input file
FITTING
J-LIST 0 1......
symmetries 1 2 .....n
itmax 10
fit_factor 100
lock 20.0
output fittest
robust 0.0
geometries c2h4_pes.dat
OBS_ENERGIES 52 (J symmetry NN Energy 12 QNs weight )
0 1 2 1343.31 0 0 0 0 0 0 0 1 0 0 0 0 10 e o
.
.
.
J-LIST is a list of total angular momentums to be included in the refinement, all checkpoint files for selected must have been already computed.
symmetries is a list of symmetries to be included, again all checkpoint files for each must have already been computed.
itmax is the number of iterations of refining carried out.
fit_factor is the relative weighting for the experimental data compared to ab initio energies. The larger this is, the more importance will be given to the experimental energies.
output is a string which specifies the pre-fix for output file names.
robust specifies whether Watson Robust fitting is used, for 0.0 it is not, for 2.0 it is.
geometries is the name of the file which contains ab initio energies. This file should give geometries in the same coordinates as specified by the potential energy surface for the molecule of interest in TROVE followed by the ab initio energy (from Molpro for example) and a weighting.
OBS_ENERGIES is the number of observed (experimental) energies used. Below this a list of energies is given in the format
J \Gamma NN E_i t_1 t_2 t_3 . . . weight e o
where and are the angular momentum and symmetry number of the energies, NN is the block number, which is the number of the energy given by TROVE. The following numbers are the TROVE assignment of the energy level, followed by a weighting.
With the fitting block added to the input, TROVE can be used to refine a PES. In the external block NPARAM should be set to the number of parameters which are to be refined. In the list of parameters, the first column of integers specifies if a parameter is to be refined. 1 will include in refinement, 0 will exclude. The next column of real numbers are the starting values of the refinement parameters and should be set to 0.0 if initial refinement.
To carry out refinement all parts of the checkpoint block should be set to read` or none`. TROVE will carry out refinement until the number of iterations specified is reached. The first iteration is essentially a checking step and does not change the value of the parameters.
Refinement Output¶
The refinement procedure produces three output files. A regular .out file with a prefix the same as the .inp file and a
.pot file and .en file with prefixes as determined by the name given in the output keyword in the Fitting block.
The main output file for refinement is straightforward. The input is repeated as with other TROVE output files and then some information is given about the eigenfunctions which were read in, etc. After this Trove prints the iteration number and then a list comparing the observed to calculated energies. For example
----------------------------------------------------------------------------
| ## | N | J | sym| Obs. | Calc.| Obs.-Calc. | Weight | K vib. quanta
---------------------------------------------------------------------------------
1 2 0 Ag 1343.5400 1346.2786 -2.7386 0.51E-03 (0) ( 0 0 0 0 0 1 0 0 0 0 0 0)*
2 3 0 Ag 1625.4000 1632.5923 -7.1923 0.26E-03 (0) ( 1 0 0 0 0 0 0 0 0 0 0 0)
3 4 0 Ag 1662.2000 1667.4972 -5.2972 0.26E-04 (0) ( 0 0 0 0 0 1 0 1 0 0 0 0)
The first number in a row is just a label to order the output. The second is the block number which was given to a particular state in the input file in the Fitting block. For the  state in the example the first energy corresponding to a
fundamental mode has a block number of 2 since 1 would correspond to the ground state with relative energy of 0. After this the angular momentum of the state, , is given along with the symmetry. The observed energy as given in the input file
is then given followed by the current iterations calculation of the energy using the adjusted potential parameters and the difference between them. The weighting given to the state is then given. The rotational
state in the example the first energy corresponding to a
fundamental mode has a block number of 2 since 1 would correspond to the ground state with relative energy of 0. After this the angular momentum of the state, , is given along with the symmetry. The observed energy as given in the input file
is then given followed by the current iterations calculation of the energy using the adjusted potential parameters and the difference between them. The weighting given to the state is then given. The rotational  quantum number and vibrational
quantum numbers are then given. If an asterisk (*) is printed at the end of the row (as in the first row of this example) it means that TROVE has assigned the state differently to how it was labelled in the input in the Fitting block.
quantum number and vibrational
quantum numbers are then given. If an asterisk (*) is printed at the end of the row (as in the first row of this example) it means that TROVE has assigned the state differently to how it was labelled in the input in the Fitting block.
TROVE then prints a list of corrections to the potential parameters followed by the new values for the potential parameters and the corrections rounded according to their error.
A table is then printed which gives details on the fit for this iteration.
-----------------------------------------------------------------------
| Iter | Points | Params | Deviat | ssq_ener | ssq_pot | Convergence |
-----------------------------------------------------------------------
| 1 | 18107 | 21 | 0.34175E-01 | 0.61230E+01 | 0.173E+03 | 0.293E+12 |
-------------------------------------------------------------------------------
This gives the statistics of the fit including both the experimental energies and the ab initio energies used to constrain the fit.
The Obs-Calc table and fit statistics is then repeated for each iteration.
The .en file gives similar information to the Obs-Calc table in the output file but gives calculated energies for all states calculated by TROVE. The .pot file is a list of the ab initio geometries with the observed (that is, the energy given for that geometry in the file listed under geometries in the Fitting bock) energies. The calculated energy is also given, which is the energy given by the potential with the corrections from refinement, along with zero-calc and the weight for the energy.