TROVE Quickstart¶
The aim of this chapter is to get a new TROVE user, especially PhD students and postdocs, up and running with the program to enable them to start carrying out calculations. It is assumed that for the molecule of interest, the molecular symmetry and geometric transforms have already been defined and that a PES and DMS has been implemented. If this is not the case then see later chapters for how this is carried out.
TROVE inputs will be treated in a general manner with occasional examples given for the phosphorus trifluoride molecule, PF3. This molecule has the same trigonal pyramidal shape as ammonia but without the complication of tunnelling inversion. This is a ‘rigid molecule’, for molecules with a non-rigid degree of freedom, see later chapters. A ‘typical’ way TROVE is used to go from vibrational calculations to full line lists is also given.
Calculation steps¶
The TROVE pipeline can broken down into the following steps:
Step 1: Hamiltonian expansion, basis sets and
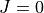 solution.
solution.Expansions of the KEO (
kinetic), PEF (potential) and dipole moment function (DMF) or any other ‘external functions’ (external) are generated and stored.Primitive 1D basis functions
 and corresponding vibrational matrix elements of expansion terms (e.g.
and corresponding vibrational matrix elements of expansion terms (e.g.  ,
,  ) -
) - basis_setand stored.Primitive rigid rotor matrix elements of the rotational angular momenta operators are constructed as stored.
A ‘contracted’, symmetry-adapted vibrational basis set is constructed (
contract) and stored.All vibrational matrix elements of the Hamiltonian operator (KEO and PEF) on the symmetry-adapted, ‘contracted’ basis functions are generated (
matelem) and stored.All vibrational matrix elements of the external operator/functions on the symmetry-adapted, ‘contracted’ basis functions are generated (
extmatelem).The
eigen-value problem is solved and the (symmetry-adapted) eigenfunctions are stored.
Step 2: Switching to the
representation (model J=0). In the following steps, the eigenfunctions are served as symmetry-adapted, contracted vibrational basis functions for the ro-vibrational problem.The
eigenfunctions are converted into the form of TROVE basis functions.The vibrational matrix elements of the (vibrational) expansion terms of the Hamiltonian and external operators are converted into the new
representation via a unitary transformation using the eigenfunctions.
Note
The pure vibrational part of the Hamiltonian is diagonal in the representation with the eigenvalues on its diagonal.
Step 3: The ro-vibrational
 eigenvalues and eigenfunctions are computed on the representation by diagonalising the Hamiltonian matrix. The latter is contracted for a given
eigenvalues and eigenfunctions are computed on the representation by diagonalising the Hamiltonian matrix. The latter is contracted for a given  as a sum-of products of the vibrational matrix elements of the Hamiltonian terms and the rotational matrix elements (Step 2) of the angular momenta (Step 1).
as a sum-of products of the vibrational matrix elements of the Hamiltonian terms and the rotational matrix elements (Step 2) of the angular momenta (Step 1).The
problem is always solved first in order to obtain the zero-point-energy (ZPE).The required
 eigenvalues and eigenfunctions are solved in any arbitrary order.
eigenvalues and eigenfunctions are solved in any arbitrary order.
The eigenfunctions are stored for further applications (e.g. intensities, PEF refinements).
Step 4: Intensity/line list calculations.
Ro-vibrational intensities are computed in for a range of the
values using the corresponding eigenfunctions and the matrix elements of the DMF from step 2 ( representation). It is a common practice to split the intensity calculations into small ranges of ![[J,J+1]](_images/math/8d75773cbe2f4a998487a31081758cc2298163cf.png) .
.
Refinement
Note
Formally, this is also Step 4, because the refinement and intensities never mix.
Note
The TROVE refinement is performed in the representation of the original (e.g. ab initio) PEF. The TROVE ‘external’ object in this case is used to represent the ‘correction’  to the ab initio PEF
to the ab initio PEF  .
.
Step 5: Preparation of the refinement of PEF. The matrix elements of the ‘external’ function (
) in the representation of the ro-vibrational eigenfunctions of the ab initio PEF are constructed (fitpot).Step 6: Refinement.
Input File Keywords and Check Point Files¶
The TROVE input file is structured in blocks with keywords within each block controlling options and parameters of the calculation. These blocks will be described individually along with a description of the checkpoint file structure. A full input file for PF3 is given at the end of the chapter.
Control block¶
For standard calculations (for relatively simple cases with no special tricks), the control block Control can be used to set the calculation step, 1, 2, 3, 4, 5, 6 as follows
Step 1:
Control
Step 1
external
end
Step 2:
Control
Step 2
end
Step 3:
Control
Step 3
J 0
end
Control
Step 3
J 1
end
Step 4:
Control
Step 4
J 0 10
end
Step 5:
Control
Step 5
end
Step 6:
Control
Step 6
end
In the following, other most important input keywords are described.
Memory Allocation¶
The total working memory which TROVE can use in a calculation is specified by
mem 20GB
where in this case TROVE could use 20 GB of RAM. If no mem command is given TROVE will assume the whole CPU memory is available which can cause problems if running on a shared memory computer. When running TROVE on such machines it is recommended that the memory is set to the combined memory of the number of cores being used.
Kinetic and Potential Energy Expansion Order¶
As described in more detail in later chapters, TROVE numerically generates the Hamiltonian using internal coordinates. The kinetic energy operator (KEO) and potential energy function (PEF) of a molecule are expanded up to a given order. The order of this expansion is specified by the keywords
KinOrder i
PotOrder j
where i and j are integers. The larger these integers are, the more accurate an expansion of the Hamiltonian is produced. For fast, test calculations, setting i and j to 2 and 4 respectively should be sufficient. For accurate results i and j should be set to 6 and 8 or 8 and 10 respectively [TROVE].
By default, the KEO is generated numerically ‘on-the-fly’ using the linearised coordinates as a Taylor-type expansion of an order  . Alternatively, the KEO can be given as a subroutine in a sum-of-products form using geometrically-defined-coordinates (referenced to as ‘local’). In this case, the corresponding expansion order is pre-defined by the given expansion.
. Alternatively, the KEO can be given as a subroutine in a sum-of-products form using geometrically-defined-coordinates (referenced to as ‘local’). In this case, the corresponding expansion order is pre-defined by the given expansion.
A possible problem which can arise here is the order of expansion of the potential energy due to the divergence of the Taylor-type expansion of the corresponding PEF. It may be that calculations are fine for low orders but on increasing the expansion order can lead to the divergence problem. This can manifest already at the stage when the primitive one-dimensional Numerov basis functions are generated (see below). It can occur if the potential expansion ‘turns over’ leading to a poor representation of the potential as shown in Figure. Remedies for this problem are increasing j to a sufficient value or reducing the range the Numerov basis is generated over. Ultimately, this problem can lead to poor or even nonphysical energy values (TROVE eigenvalues).
Number of Atoms and Modes¶
The number of atoms and vibrational modes is defined by the keywords
Natoms N
Nmodes i
where i is normally  for non linear molecules and
for non linear molecules and  for linear molecules.
for linear molecules.
Primitive Block¶
The polyad number and maximum energy for the primitive basis functions are set in the primitive block:
PRIMITIVES
Npolyads p
enercut x
END
As discussed in more detail in later chapters, TROVE uses products of primitive one-dimensional functions as basis functions. The polyad number,  , is a way to restrict the size of the basis set [TROVE]. The sum of the primitive function’s vibrational quantum number,
, is a way to restrict the size of the basis set [TROVE]. The sum of the primitive function’s vibrational quantum number,  , is then restricted to be below
, is then restricted to be below N, that is:
(1)¶
where  is an integer used to control the basis further. For fast, test calculations a low value of
is an integer used to control the basis further. For fast, test calculations a low value of  should be used, for example 4.
Increasing n gives a larger basis set which will give more accurate results (and extend the energy range) at the expense of time and memory.
Note that because of how the polyad number is defined, even increasing by 1 can lead to a much larger basis.
should be used, for example 4.
Increasing n gives a larger basis set which will give more accurate results (and extend the energy range) at the expense of time and memory.
Note that because of how the polyad number is defined, even increasing by 1 can lead to a much larger basis.
Another way of limiting the size of the primitive basis functions is by energy. The enercut keyword specifies the maximum energy a primitive basis function can have if it is to be included. As discussed below, the basis set is further restricted by energy in the Contraction block and so usually x is set to a large value. For example 40,000 (in units of wavenumbers, cm-1).
Contraction Block¶
As will be discussed in later chapters, TROVE builds contracted basis functions from the primitive basis functions. In the Contraction block the parameters for making this contraction are chosen. A Contraction block example is
CONTRACTION
Npolyads n
enercut x
coeff_thresh 1.0d-30
degeneracy 1d-02
sample_points 40
sample_attempts 500
symm_toler 1.0d-3
END
Npolyads is the same as for the Primitive block and defined in equation (1) and sets the maximum polyad number for the contraction.
Similar to the primitive block, enercut sets the maximum energy a contraction can have. The value of enercut chosen depends on the application.
Because the maximum energy of the contracted basis functions are set, it makes sense to set the value of enercut in the Primitive block large.
Also given in the Contraction block are parameters relating to how TROVE works out the symmetry of contracted basis functions (coeff_thresh, degeneracy). This will be described in later chapters and has been discussed in a recent publication [17YuYaOv].
Symmetry¶
The symmetry of the molecule is specified by the SYMGROUP keyword. The symmetry of a given molecule is set in the mol_*.f90 file which, as ever, will be discussed in later chapters. For PF3 the SYMGROUP is set using
SYMGROUP C3v(M)
Diagonalizer Block¶
The Diagonalizer block determines the way in which the Hamiltonian matrices are diagonalized. The method of carrying out the diagonalization is specified by a keyword related to the LAPACK/BLAS program which are used. These are standard programs used for carrying out matrix manipulations used in many areas of science, engineering, mathematics, etc. SYEV (equivalent to DSYEV) is the default value which computes all eigenvalues and eigenvectors. SYEVR allows an uppervalue on the computed eigenvalues to be specified. There is another keyword, enermax, which limits the energies of eigenfunctions which are saved. For example
DIAGONALIZER
SYEV
enermax 16000.0
end
If a pure vibrational calculation ( ) is being carried out, the energies of excited states are automatically given relative to the zero point energy (ZPE) of the ground vibrational state. For
) is being carried out, the energies of excited states are automatically given relative to the zero point energy (ZPE) of the ground vibrational state. For  calculations, the ZPE value is taken from an checkpoint file containing the energies, see below. The keyword
calculations, the ZPE value is taken from an checkpoint file containing the energies, see below. The keyword ZPE followed by the vibrational zero point energy can be specified explicitly so that rotational-vibrational energies are also given relative to any desired value, e.g. 0.000.
For large calculations, it is more efficient to diagonalize each symmetry’s Hamiltonian matrix separately. The symmetry of interest is specified using the keyword gamma n where n = 1,2.. is the symmetry of interest, e.g. for the second irreducible representation of a given group:
DIAGONALIZER
SYEV
gamma 2
end
Print Out Level¶
The amount of output printed is specified by the verbose keyword. A value of 4 is sufficient for most purposes.
verbose 4
Increasing this value will produce more output, this is useful for debugging, etc.
Specifying the Molecule¶
The molecule is defined in TROVE by the following
dstep 0.01
COORDS linear
TRANSFORM r-alpha
MOLTYPE XY3
MOLECULE PF3
REFER-CONF RIGID
dstep has to do with how fine TROVE carries out the finite derivatives when the PEF or DMF is re-expanded in terms of the internal TROVE coordinates.
The COORDS keyword specifies the type of internal coordinates. The standard option is linear which indicates that the kinetic and potential energy should be expanded in linear coordinates [TROVE]. Another option is local which uses curvilinear (geometrically defined) coordinates [15YaYu]. For the curvilinear coordinates, the KEO operators are currently available for a few specific cases. Externally generated curvilinear KEOs can be also used, provided they are given in some TROVE-standard form.
TRANSFORM specifies how to transform the coordinates from Z-matrix to the coordinates used in TROVE. This is specified in the mol_*.f90 file for the molecule of interest. For the PF3 example here, the details of the transformation are given in the r-alpha subroutine.
As the symmetry transforms only need to specified for each type of molecule of the same symmetry, they can be reused. For example PCl3 belongs to the same symmetry group as PF3. The MOLTYPE keyword identifies the “type of molecule” and molecules of the same symmetry can then be straightforwardly used. This keyword specifies the subroutine to use to define rotational symmetries, etc.
MOLECULE is an optional keyword which specifies the molecule’s name.
Whether the molecule is rigid or non-rigid is specified with the REFER-CONF keyword. For non-rigid molecules a special degree of freedom which is large amplitude (or ‘floppy’) can be specified. Examples include the inversion motion in ammonia or the torsional motion in ethane. In this case HBJ theory (see Theory chapter) can be used. These rigid or non-rigid reference frames are used as expansion centres to represent PEF, KEO and DMF in the form of sum-of-products.
Z-Matrix Block¶
TROVE uses Z-matrix coordinates as intermediates to define the actual internal coordinates. To this end, the following Z-matrix block is used to specify the molecule’s geometry as well as the masses of atoms. For example for PF3 the Z-matrix is
ZMAT
P 0 0 0 0 30.973761998
F 1 0 0 0 18.998403162
F 1 2 0 0 18.998403162
F 1 2 3 0 18.998403162
end
The Z-matrix used by TROVE is very similar to those used by electronic structure programs such as Molpro and Gaussian. The first column is the atom’s (element) symbol. The second column is the atom which the atom of that row is connected to. The third column is the bond angle between the atom of the row and a specified atom. The fourth column is the dihedral angle between the atom of that row and a specified atom. The fifth column has to do with the way a particular molecule type is set up in TROVE and describes the type of dihedral angle. The sixth column is the atom’s mass in atomic mass units. Note that isotope masses should be used, not averaged atomic weights.
Basis Block¶
The Basis block specifies the type of basis functions used by TROVE and how the kinetic and potential energy is expanded for each coordinate. Specifically, the one-dimensional basis functions which will then be used to build up contracted and symmetrized functions. An example for PF3 is
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear','morse',range 0,7, weight 2.0, points 2000, borders -0.4,2.0
1,'numerov','linear','morse',range 0,7, weight 2.0, points 2000, borders -0.4,2.0
1,'numerov','linear','morse',range 0,7, weight 2.0, points 2000, borders -0.4,2.0
2,'numerov','linear','linear' range 0,14, weight 1.0, points 2000, borders -1.3,1.3
2,'numerov','linear','linear',range 0,14, weight 1.0, points 2000, borders -1.3,1.3
2,'numerov','linear','linear',range 0,14, weight 1.0, points 2000, borders -1.3,1.3
END
The first line in this block, 0, JKtau, Jrot 0 specifies the rotational functions. For  calculations the value of
calculations the value of Jrot is changed to of interest. PF3 has  internal degrees of freedom and thus 6 basis functions are required. Basis functions are grouped using an integer label. For this example, ‘1’s are the P-F stretches and ‘2’s are the P-F bends. The grouping is used for producing symmetric
combinations of basis functions and only coordinates symmetrically related should be grouped together. Details of this procedure are discussed in Chapter Theory and in [17YuYaOv].
internal degrees of freedom and thus 6 basis functions are required. Basis functions are grouped using an integer label. For this example, ‘1’s are the P-F stretches and ‘2’s are the P-F bends. The grouping is used for producing symmetric
combinations of basis functions and only coordinates symmetrically related should be grouped together. Details of this procedure are discussed in Chapter Theory and in [17YuYaOv].
For a given basis function row the options are as follows. The first keyword specifies what the one-dimensional basis functions are. In this example they are numerically generated using the Numerov-Cooley method. Other options are harmonic and morse where these analytical basis functions shall be used. The second keyword specifies how the kinetic energy operator is expanded. The third keyword gives the expansion coordinates for the potential. Here ‘Morse coordinates’ of the form  are used for the stretching coordinates while
are used for the stretching coordinates while linear (the angles themselves) coordinates are used for the bends.
The numbers after range specify the range of vibrational quantum numbers of the one-dimensional functions to be used. For the example here, 0-7 is used for stretches and 0-14 for bends. This is related to the definition of the maximum polyad number used in equation (1). The number after weight gives the weighting of the vibrational quantum number for that coordinate in equation (1). Since the P-F stretches here have a waiting of 2, it only makes sense to generate them from 0-7 if the polyad number is set to 14. The legacy aliases for weight are resc (resonance coefficients).
points and borders specify the number of points and the starting points for the Numerov-Cooley integration. Generating these one-dimensional functions is fast and so many points should be taken. The borders should be set far enough into the classically forbidden region of the potential such that the results are not sensitive to slightly larger or lower values. The units for borders are the same as those used that the potential was expanded in (Morse for stretches and angles in radians for bends in this example).
The details of the primitive basis sets are given in the TROVE output file and will be discussed in Chapter Outputs.
Checkpoint Block¶
The Checkpoint block determines which checkpoint files are saved by TROVE. This is an important aspect of TROVE as usually calculations are built up sequentially. The checkpoint files allow a calculation to be restarted with the results of previous calculations read in by TROVE. For each keyword in the Checkpoint block the options are read or save. If read is specified then the checkpoint file (.chk) associated with that keyword must be present in the directory where the calculation is run. In this case that file will be read in for TROVE to use. If save is specified then the checkpoint file associated with that keyword will be saved.
The kinetic.chk and potentail.chk contain the expansion coefficients of the KEO and PEF expansions, controlled by the Kinorder and Potorder keywords discussed above. The associated keywords are kinetic and potential, respectively. The corresponding input values are save, read or none. This is usually the first part of a TROVE calculation. Once these objects are generated with the expansions to a sufficient order (for example 6/8 for kin/pot order) it can be reused while different basis sets, polyads, etc are compared.
Example:
check_point
kinetic save
potential save
external save
....
end
If transition moments or intensity calculations are being carried out then the keyword external should be included and set to save. This generates an expansion of the DMF and requires a DMF to be provided.
The primitive basis set can be saved/read with the basis_set keyword. This will generate .chk files with the one dimensional numerov and contracted primitive basis functions. The corresponding data with the primitive basis functions and 1D matrix elements are stored in two checkpoint files,
numerov_bset.chk and prim_bset.chk.
The contracted, symmetry-adapted basis is saved/read with the contract keyword and generates a contr_vectors.chk checkpoint file and human readable files contr_descr.chk and contr_quanta.chk. For example,
check_point
...
basis_set save
CONTRACT save
....
end
The matrix elements of the Hamiltonian between contracted functions can be saved using the matelem keyword. The file contr_matelem.chk is generated. This can be very large depending on the basis set. There is an option to split this checkpoint file into 13 files responsible for different parts of the Hamiltonian operator (see below). in this case, contr_matelem.chk`` contains matrix elements of the pure vibrational part of the Hamiltonian, while other parts are stored in matelem_1.chk, matelem_2.chk … matelem_12.chk. To use the option, the keyword split should be added, which can be followed by the numbers specifying the term to compute, e.g. to read all checkpoints of the KEO matrix elements in the split form:
check_point
...
matelem read split
extmatelem none
....
end
or to compute the terms from 1 to 6:
check_point
...
matelem save split 1 6
extmatelem none
....
end
Similarly, vibrational elements of the DMS can be saved using the keyword extmatelem to generate the contr_extfield.chk file, which can also be split into sub-parts (3 for the 3D dipole moment object).
If the eigenfunction of the calculation are required (for example for transition moment calculations) then the EIGENFUNC keyword should be set to save.
This generates eigen_vectors[J].chk files and human readable eigen_descr[J].chk files, where J is the rotational quantum number. The eigenfunctions are used to for generating basis functions for calculations as discussed below, e.g.
check_point
....
EIGENFUNC save
....
end
Note
The Step 1 in the control block
- ::
- control
Step 1
end
corresponds to and replaces the following configuration of the
check_pointblock:
- ::
check_point kinetic save potential save basis_set save CONTRACT save matelem save eigenfunc save end
In this case the check_point block should be omitted. However a combination of the control and check_point blocks can be used to specify a non-standard configuration. In this case the control block must appear first.
A description of how these files are used for calculations is given below.
Equilibrium Block¶
The Equilibrium block specifies the equilibrium bond lengths (in Angstrom) and bond angles of the molecule. TROVE uses these values to calculate Cartesian coordinates and transform between coordinate systems. For PF:math:_3 this is
EQUILIBRIUM
Re 0 1.56
Re 0 1.56
Re 0 1.56
alphae 0 98.000 deg
alphae 0 98.000 deg
alphae 0 98.000 deg
end
Specparam Block¶
The Specparam block is used to define special parameters. For example, the value of  in the Morse potential
function.
in the Morse potential
function.
Poten Block¶
The Poten block is used to specify the PES. For PF:math:_3 the first few lines are
POTEN
NPARAM 304
POT_TYPE poten_xy3_morbid_10
COEFF list (powers or list)
VE 0 0.000000000000
FA1 1 -5730.010012350451
FA2 1 1091683.728331340943
FA3 1 -1947258.254744407022
.
.
.
NPARAM is used to specify the number of parameters used to define the PES. POT_TYPE is the name of the potential energy surface being used which is defined in the pot_*.f90 file. The COEFF keyword specifies whether the potential is given as a simple list or if the powers or the expansion are given. This depends on how the potential has been set up. The list of PES parameters is then given.
External (Dipole) Block¶
The External block is similar to the Potential block but defines other functions to be included in the calculations. Most commonly this will be the dipole moment surface (DMS). For example for PF:math:_3 the first few lines are
DIPOLE
DIMENSION 3
NPARAM 127 0 0
DMS_TYPE XY3_MB
COEFF list
dstep 0.005
COORDS linear
Order 6
parameters
charge 0 0.0
order 0 4.0
alphae 0 98.000000000000
re14 0 1.560000000000
beta 0 1.000000000000
gamma 0 0.000000000000
delta 0 0.000000000000
mu0 1 -0.177517341983
F1 1 -2.287669265640
....
....
end
As the DMS is a vector function (it has values for the x, y and z directions) the three numbers of parameters for each is specified in nparam. For PF:math:_3 only one direction is needed however due to the way the DMS is specified. The name of the DMS is specifed by DMS_TYPE which corresponds to the name in the .mol file. COEFF specifies how the parameters are given (a list in this case) and COORDS is used to describe which coordinates are used to expand the dipole in TROVE. Order specifies the order to expand the dipole to, similar to the keywords for the kinetic and potential energy. The list of parameters is then given in a similar way to the Poten block.
The external block is also used to refine potential energy surfaces as discussed in the Refinement chapter. It can even be used for more exotic applications such as introducing quadrupole potentials, etc but this will not be covered here.
Intensity Block (Step 4)¶
As described below, once eigenfunction for the vibrational and rotational states are calculated, they can be used to calculate the intensity of transitions (step 4). Options for controlling this in TROVE are specified in the Intensity block.
Transition moments (TMs) can be calculated once vibrational () eigenfunctions are available (see below). In this case the Intensity block is given, for example
INTENSITY
tm
THRESH_TM 1e-12
ZPE 11014.221565
selection (rules) 1 1 1 1 1 1 1 1 (N of irreps)
J, 0,0
freq-window -0.0001, 5000.0
energy low -0.0001, 2000.0, upper -0.00, 7000.0
END
tm tells TROVE to calculate transition moments only. THRESH_TM sets the threshold for the smallest TMs to be calculated. ZPE is the value of the molecule’s zero-point energy.
For calculating absorption intensities the Intensity block takes the following form
INTENSITY
absorption
THRESH_INTES 1e-20
THRESH_LINE 1e-20
THRESH_COEFF 1e-18
TEMPERATURE 300.0
Partition 1000.0
GNS 8.0 8.0 8.0
selection (rules) 1 1 2 (N of irreps)
J, 0,10
freq-window -0.1, 4000.0
energy low -0.1, 2000.0, upper -0.1, 6000.0
END
absorption specifies that absorption intensities between states are to be calculated.
THRESH_INTES/LINE/COEFF are used to control the level of print out for intensities. Very large outputs can be produced if these are set very low (as needed for ‘production’ quality line lists) but for quicker checks higher values should be used.
TEMPERATURE is used to specify the temperature of interest. This will affect the population of states (Boltzmann population).
Partition is the value of the partition function. This can be calculated from all of the ro-vibrational energy levels used. Note that at high temperatures enough energy levels must be included for accurate results. If this is not the case (for example, for a test calculation) then a literature value could be used.
GNS is the spin statistical weights for each symmetry. These can be looked up for many molecules or worked out from the procedure in Bunker and Jensen, chapter 8 [98BuJe]. selection is used to specify which symmetries can make up the initial and final states of a transition. The product of the upper and lower eigenfunctions must contain a component of the dipole itself [98BuJe]. Thus for the PF3 example,  and
and  are grouped together while E can only go to E. Integers are used to form groups, in this case 1 1 are for and and 2 is for E.
are grouped together while E can only go to E. Integers are used to form groups, in this case 1 1 are for and and 2 is for E.
J, i,j specifies the rotational states to be included. In the example above 0 to 10 were used. It is often better to split a calculation into 0,1-1,2-2,3, etc to fit into time allocations on computers. The vibrational states to be included can also be specified by the v i, lower x, y, upper x', y'
where i is the number of a vibrational mode and x, x’ and y, y’ give the limits for the lower and upper states included. If this is not included then all vibrational states are considered.
freq-window This specifies the frequency window (in wavenumbers) in the spectra to be used. In the example here -0.1 is used as the minimum to guarantee values from 0 are used while 4000 is the maximum considered. energy low specifies the energies of the lower and upper states to be included. In the example the highest energy lower state to include it 2000 so since the maximum frequency of light considered is 4000, the upper state needs a maximum of 6000 (energy proportional to frequency,  ).
).
To calculate absorption intensities the eigenfunctions and eigenvalue files of the states to be included must be included in the directory where TROVE is run. More on this will be described below.
The working equations for intensity calculations are discussed in the Theory chapter.
ExoMol Line list calculations¶
In order to produce a line list in the ExoMol format, the keyword exomol must be added anywhere inside the intensity block:
Intensity
....
exomol
name *filename*
...
end
where filename defines the name of the line list files (.states and .trans). In this case, the absorption intensities computed for the temperature and partition functions specifies are not printed out only used together with the corresponding threshold THRESH_INTES to decide to keep the line in the line list or not.
Practical Guide to Running TROVE¶
In this section the recommended steps for using TROVE are described, from calculating vibrational energies up to rotational-vibrational absorption intensities. It will be assumed that the PES and DMS are available and that the symmetry group, Z-matrix, primitive basis set, etc have been set up. These inputs are generally fixed once they have been decided on and typically the user does not need to modify them.
This section can be followed most easily in conjunction with the TROVE training directory which should come with this manual. This contains a TROVE executable file and inputs, outputs and checkpoints for a model PF3 calculation as well as a README file. It may be necessary to compile a version of TROVE on the local computer to get working executable.
The first step in any TROVE calculation is the production of the hamiltonian.chk checkpoint file. As discussed above, this contains the details of the kinetic and PES expansion and if required, the DMS expansion, which are used in later parts of TROVE. In the Checkpoint block the following should be set to save
check_point
kinetic save
potential save
external save
....
end
This will generate the hamiltonian.chk file which will be read in subsequent calculations. The time taken and memory usage of this step can vary depending on the expansion orders of the kinetic energy, PES and DMS. As mentioned above, low expansion orders (for example 2 and 4 for kinetic and potential respectively) are useful for test calculations but are not very accurate but larger expansions (e.g 6 and 8) take a longer time to compute and use.
The basis set checkpoint files are usually generated next. In the Checkpoint block this is specified by
check_point
...
basis_set save
CONTRACT save
....
end
The basis_set keyword generates the file prim_bset.chk and, if a Numerov basis is selected, numerov_bset.chk. CONTRACT generates the file contr_vectors.chk which contains the contracted basis functions. This also generates the file contr_matelem.chk which contains vibrational matrix elements of the Hamiltonian in the contracted basis representation. Depending on the size of the basis set, this file can be very large. The human readable files contr_descr.chk and contr_quanta.chk are also generated which contain descriptions of the contracted basis functions and of the energies corresponding to the contracted basis functions.
At this stage, TROVE will calculate and output the vibrational energies. The eigenfunctions for each vibrational state are saved using
check_point
....
EIGENFUNC save
....
end
These are used in subsequent transition moment and absorption intensity calculations. A series of files, eigen_vectors0_n.chk are generated where n ranges from 1 to however many symmetry classes there are for the molecule of interest. Similar to the contracted basis, eigen_desc0_n.chk human readable files for each symmetry class of eigenvectors are also generated along with eigen_quanta0.chk which contains a description of eigenvectors and eigenvalues.
The steps described above can all be carried out with a single run of TROVE, setting all of the keywords to save. For large calculations however, it is usually best to build up the checkpoint files, checking each step is successful. To follow the steps outlined above, the keywords should be set to read for .chk files which have already been generated. For example, once the hamiltonian.chk file is generated, kinetic and potential can be set to read.
Once the contr_matelem.chk file has been created along with vibrational eigenfunctions, it is in principle possible to calculate J:math:>0 energies.
A faster and more efficient way to do this however is to make use of the J=0 representation. This is where the vibrational eigenfunctions for J=0 calculation are used as a basis set for J:math:>0 calculations. This usually leads to much faster calculations of excited rotational states. To use this method put model j=0 anywhere in the Contraction block and in the Checkpoint block put
check_point
.....
contract save
matelem convert
extmatelem convert
eigenfunc read
end
This will produce a new file, j0_matelem.chk and, if extmatelem specified, j0_extfield.chk. j0contr_descr.chk, j0contr_quanta.chk and
j0contr_vectors.chk files are also generated, equivalent to those described above. A calculation should then be run setting CONTRACT and matelem to read and EIGENFUNC save. This will produce a desc and checkpoint files for the eigenfunctions but saved in the J=0 representation.
Once these files have been generated it is then straightforward to carry out calculations for . In the Basis block change
0,JKtau, Jrot 0
to
0,JKtau, Jrot 1
(or whatever J of interest). The model j=0 keyword should be left in the Contraction block. In the Diagonalizer block the keyword ZPE should be added to set the vibrational zero point energy. The ro-vibrational energy levels will then be given with respect to this. In the Checkpoint block everything should be set to read apart from EIGENFUNC if the rotational eigenfunctions are required.
Transition moments can be calculated by inserting the Intensity block into the input file as described above. The directory in which TROVE is run should contain the vibrational eigenfunctions stored either in the standard contracted form (eigen_vectors0.chk) or the J=0 form (J0eigen_vectors0.chk).
Absorption intensities (line lists) can be calculated once the rotational-vibrational eigenfunctions of interest have been calculated, usually using the J=0 method. The relevant .chk files describing the eigenfunctions should all be present in the directory where TROVE is run. The Intensity block should be included in the input block with the absorption keyword as described above.
For both transition moment and absorption intensity calculations everything should be set to read in the Checkpoint block (with the relevant checkpoint files included in the directory).
Although TROVE can calculate intensities, the GPU program GAIN can do this far faster [GAIN]. The use of the program will be described in Chapter _linelists but the input is the same as described above.
Sample TROVE Input File¶
Below is a sample TROVE input file for the molecule PF3. Using this file (and adding in Intensity blocks when needed) a full line list for this molecule could be produced. To save space the PES and DMS parameters have not been included in full. The actual text file should be kept in the same directory as this manual.
mem 20 gb
KinOrder 6 (Max order in the kinetic energy expansion)
PotOrder 8 (Max order in the potential energy expansion)
Natoms 4 (Number of atoms)
Nmodes 6 (Number of modes = 3*Natoms-6)
Control
step 1
end
(ACTIVE SPACE CUTOFFS:)
PRIMITIVES
Npolyads 14 (how many polyads we calculate)
enercut 100000.(energy cut in the primitive matrix for the diagonalization)
END
CONTRACTION
Npolyads 14 (how many polyads in the contracted represent.)
enercut 100000. (energy cut in the primitive matrix for the diagonalization)
degeneracy 1e-3 (threshold to define degeneracy)
sample_points 40
sample_attempts 500
symm_toler 1e-3
coeff_thresh 1e-16
exp_coeff_thresh 1.0d-8
END
verbose 3
DIAGONALIZER
SYEV
end
dste p 0.01 (finite difference element for each mode )
TRANSFORM r-alpha
MOLTYPE XY3
MOLECULE PF3
COORDS linear
REFER-CONF RIGID (Reference configuarion: RIGID or NON-RIGID)
SYMGROUP C3v(M)
ZMAT
P 0 0 0 0 30.973761998
F 1 0 0 0 18.998403162
F 1 2 0 0 18.998403162
F 1 2 3 0 18.998403162
end
BASIS
0,'JKtau', Jrot 0
1,'numerov','linear','morse',range 0,7,resc 2.0,points 2000, borders -0.4,2.0
1,'numerov','linear','morse',range 0,7,resc 2.0, points 2000, borders -0.4,2.0
1,'numerov','linear','morse',range 0,7, resc 2.0, points 2000, borders -0.4,2.0
2,'numerov','linear','linear',range 0,14,resc 1.0, points 2000, borders -1.3,1.3
2,'numerov','linear','linear',range 0,14,resc 1.0, points 2000, borders -1.3,1.3
2,'numerov','linear','linear',range 0,14,resc 1.0, points 2000, borders -1.3,1.3
END
EQUILIBRIUM
Re 0 1.56
Re 0 1.56
Re 0 1.56
alphae 0 98.000 deg
alphae 0 98.000 deg
alphae 0 98.000 deg
end
SPECPARAM
beta 0 1.00000
beta 0 1.00000
beta 0 1.00000
END
POTEN
NPARAM 304
POT_TYPE poten_xy3_morbid_10
COEFF list (powers or list)
VE 0 0.000000000000
FA1 1 -5730.010012350451
FA2 1 1091683.728331340943
FA3 1 -1947258.254744407022
FA4 1 18286059.212070591748
FA5 1 -105327110.803434416652
.
.
.
.
end
DIPOLE
rank 3
NPARAM 127 0 0
DMS_TYPE XY3_MB
COEFF list
dstep 0.005
COORDS linear
Order 6
parameters
charge 0 0.0
order 0 4.0
alphae 0 98.000000000000
re14 0 1.560000000000
beta 0 1.000000000000
gamma 0 0.000000000000
delta 0 0.000000000000
mu0 1 -0.177517341983
F1 1 -2.287669265640
F3 1 0.432166856494
F4 1 -0.037093470208
F5 1 -0.761988732763
.
.
.
.
.
end
As a more sophisticating alternative to the Control block, the following Checkpoint structure can be used to specify the individual calculation options at Step 1
CHECK_POINT
ascii
kinetic read
potential read
external none
basis_set save
CONTRACT save
matelem save split
extmatelem none
EIGENFUNC none
END